
Syed Rahman
“Our results demonstrate an unsuspected link between the cancer-causing abilities of some cells and abnormal circadian and metabolic rhythms, which we surmise to be advantageous for cancer.” -Chi Van Dang (Director of the Abramson Cancer Center)
Cancer is defined as essentially uncontrolled cell division in abnormal human cells. The current treatment for many cancers is attempting to physically remove the cancer cells that have formed—what are seen as tumors, large and dense cell masses. Additionally, cocktails of chemotherapy treatments are also used including cytotoxic drugs such as doxorubicin. However, since these treatment options are very dangerous for the patient (such as from killing healthy cells) and are not completely remissive of the cancer, a great deal of research is being done into trying to find other molecular targets to target via drugs. Moreover, these molecular targets are simply proteins or genes in cancer cells that play a role in cellular processes that impact or are impacted by certain cancers. Therefore, it is believed that these proteins and genes can be targeted via drugs to indirectly impact the cancer itself or at least its symptoms. C-myc is a gene that has been researched and found to impact a variety of cellular process that cancer impacts or impacts cancer, such as cellular aging, cellular biorhythms, programmed cell death, and even interactions with other cancer causing genes in the cell as a whole.
C-myc is a protein transcription factor that has the ability to activate and repress target genes in the genome. Specifically, it is a basic helix-loop-helix leucine zipper transcription factor. It was discovered as the homolog of an oncogene associated with avian retroviruses and has been the subject of many studies involving a variety of cellular processes such as growth control and cellular differentiation. Recently, c-myc has been widely researched in a variety of cancers (Hoffman & Liebermann 2008). This is because many of its target genes control cellular processes that are commonly affected in cancers, such as cell cycle control and protein synthesis. Moreover, it has been shown that a variety of human cancers coincide with chromosomal abnormalities that in term deregulate c-myc protein, thus showing a possible way to regulate its levels in the body (Hoffman & Liebermann 2008). Overall, since c-myc can impact cancer in a variety of ways inside the cell (such as being important in apoptosis) and can be regulated, it seems to be an interesting topic to investigate as a potential molecular target.
C-myc functions as a transcriptional regulator by binding to a basic helix-loop-helix protein called MAX. Other partners such as Mx1 and MAD join c-myc as well, but MAX remains the most significant member of the complex that forms with c-myc. This c-myc and MAX complex eventually interacts with transcriptional coactivators and induces repression of a variety of other genes (Hoffman & Liebermann 2008). This complex is known to have a binding site on the p21 core promoter, another oncogene in cancer(Hoffman & Liebermann 2008). Therefore, it seems possible that c-myc can regulate other genes that directly affect other cancers. This could potentially be taken advantage of in the clinical setting.
C-myc has also exhibited a severe impact on cellular apoptosis, or programmed cell death. The study done by Askew employed myeloid progenitor cells which depend on interleukin 3 (IL-3) to grow and survive. When IL-3 was withdrawn from the cells, the level of c-myc was downregulated and cells became arrested in the G0 (or resting) phase of the cell cycle. These cells eventually underwent apoptosis (Askew Et. Al. 1991). Additionally, it was also found that enforced c-myc expression accelerates the apoptotic response as compared to IL-3 withdrawn cells that did not have enforced c-myc expression (Askew Et. Al. 1991). Thus, there is a correlation between c-myc levels and cellular death. This has been shown to happen through crosstalk with another gene present in many cancers called p53, a tumor suppressor gene which causes apoptosis in response to many different types of stimuli. C-myc has been shown to induce expression of the ADP ribosylation factor (ARF) protein, which will in turn increase the amount of p53 protein in the cell. Once again we see a crosstalk between c-myc and another gene that is important in regulating cancer, but also the apoptosis or death of cancer cells. This identifies c-myc as a critical gene in between other cancer causing genes that can be regulated externally to potentially even go as far as to kill cancer cells. Thus, it seems very clear that c-myc has a potential role in mediating a variety of other target genes in cancers that are known to have some role in cellular apoptosis and cell cycle state (Hoffman & Liebermann 2008).
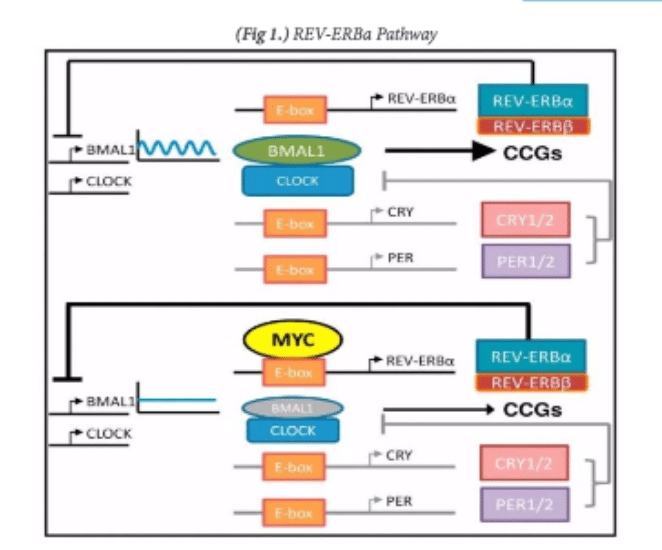
Thus, there is oncogenic potential for c-myc as a target to directly impact a variety of pathways involved in oncogenesis. Additionally, it has also been shown to have an important role in impacting normal 24- hour rhythms in cancer cells, called circadian rhythms (University of Pennsylvania School of Medicine). It has recently been found that c-myc cancers have altered circadian rhythms in cells through the upregulation of the “clock” protein known as REV-ERBa (Altman et al., 2015) [Figure 1]. This lowers the expression of another protein known as BMAL1.
It is interesting because it has been shown that higher REV-ERBa expression and lower BMAL1 expression predicts worse clinical outcome for neuroblastoma patients in a clinical study done in The Children’s Hospital of Philadelphia and the University of Cologne in Germany with a sample size of survival data from over 740 patients (University of Pennsylvania School of Medicine 2015). Neuroblastomas are a very malignant tumor made up of neuroblast cells usually found on the adrenal glands of the kidney. This has the potential of being very useful in treating these cancers since they have a very poor clinical outcome. The potential of using c-myc as a molecular target is seen from the unusual circadian rhythms to affect the amount of REV-ERBa protein in the cell, which has been shown to affect neuroblastoma patient prognosis through some unknown mean (Altman Et. Al. 2001). Overall, there seems to be some potential in impacting clinical performance by affecting the role that c-myc has in various pathways.
Finally, c-myc has also been shown to affect the formation of telomeres in human cells. It has been shown that c-myc expression stimulated telomerase activity in human mammary epithelial cells that normally lack it. This leads to the possibility that one of the reasons that c-myc is prone to cause tumors and be deregulated in a variety of cancers is its ability to induce telomerase since tumor cell lines are almost all telomerase positive (Wang Et. Al. 1998). This just leads to another example of how it is possible for c-myc to affect the cell in a way that would in turn affect the progression of cancers. If this can be taken advantage of clinically, cancer treatments might be significantly improved.
Overall, c-myc has the potential to be investigated as a target for many different pathways in cancer. Its role in telomerase induction, circadian rhythms, and interplay with other cancer causing genes suggests ways for this gene to impact cancers. It is possible that drugs can affect the expression of the c-myc gene in different ways (such as overexpression or underexpression) in order to impact these roles and potentially treat certain cancers in a more effective way. However, much more research needs to be done in order to see the efficacy of c-myc as a potential target in treating cancers and identifying the particular type of repressor or drug that will affect its expression.